黄卓团队Advanced Science合作揭示PTPRN通过促进NaV1.2通道内吞发挥癫痫保护作用新机制
神经可塑性是神经系统快速适应内外环境变化的基本特性,分为突触可塑性和内在可塑性[1]。突触可塑性通过调整突触连接精细化神经网络,而内在可塑性通过改变离子通道的数量、分布和活动,调节神经元膜的内在兴奋性[2,3,4]。癫痫是一种以大脑中异常和过度神经活动为特征的神经疾病,内在可塑性在癫痫活动的生成、传播及其抑制中起关键作用[5,6]。离子通道的表达和分布变化不仅促进癫痫的发展,也有助于限制其发作和防止癫痫进展。然而,目前对于内在可塑性和癫痫发生背景下的离子通道运输机制了解甚少,需要进一步研究。
近期,北京大学药学院、IDG麦戈文脑科学研究所黄卓教授和重庆医科大学附属第一医院田鑫教授研究团队共同在Advanced Science期刊发表题为“A Novel Ubiquitin Ligase Adaptor PTPRN Suppresses Seizure Susceptibility through Endocytosis of NaV1.2 Sodium Channels”的研究论文。这项研究通过加权基因共表达网络分析(WGCNA)发现N型蛋白酪氨酸磷酸酶样受体(PTPRN)在调节颞叶癫痫神经元兴奋性中起关键作用。研究揭示增强的神经元活动增加了PTPRN的表达,PTPRN通过招募NEDD4样E3泛素蛋白连接酶(NEDD4L)到NaV1.2钠通道,促进其泛素化和内吞,平衡异常的神经元兴奋性和癫痫易感性。研究阐明了PTPRN在调控NaV1.2功能中的独特机制,为癫痫等神经疾病的治疗提供了新的策略和方法。

颞叶癫痫(TLE)是一种复杂的癫痫类型,其发病机制尚未完全明晰。为了更好地理解TLE的潜在机制,研究人员利用WGCNA分析了大鼠海马颗粒细胞的RNA测序数据,构建基因共表达网络,识别出与癫痫表型显著相关的基因模块。在筛选的候选基因中,PTPRN在两个TLE模型中均显著表达(图1 A-C)。进一步分析TLE患者的颞叶组织样本发现,TLE患者的PTPRN蛋白水平显著高于非TLE患者(图1 D)。同样,在KA和pilocarpine动物模型中,PTPRN的蛋白和mRNA水平在癫痫诱导后显著上升(图1 E,F)。免疫组织化学分析显示,PTPRN表达主要集中于DG,并且在KA处理后表达增加(图1 G-I)。原代神经元的实验表明,KCl诱导的膜去极化导致PTPRN mRNA和蛋白水平增加,而通过钠通道阻滞剂TTX抑制神经活动,PTPRN表达水平下降(图1 J,K)。这些结果表明,PTPRN在TLE中表达水平异常升高,并且其表达受到神经活动的调节,暗示PTPRN可能在TLE相关的神经可塑性中发挥重要作用。
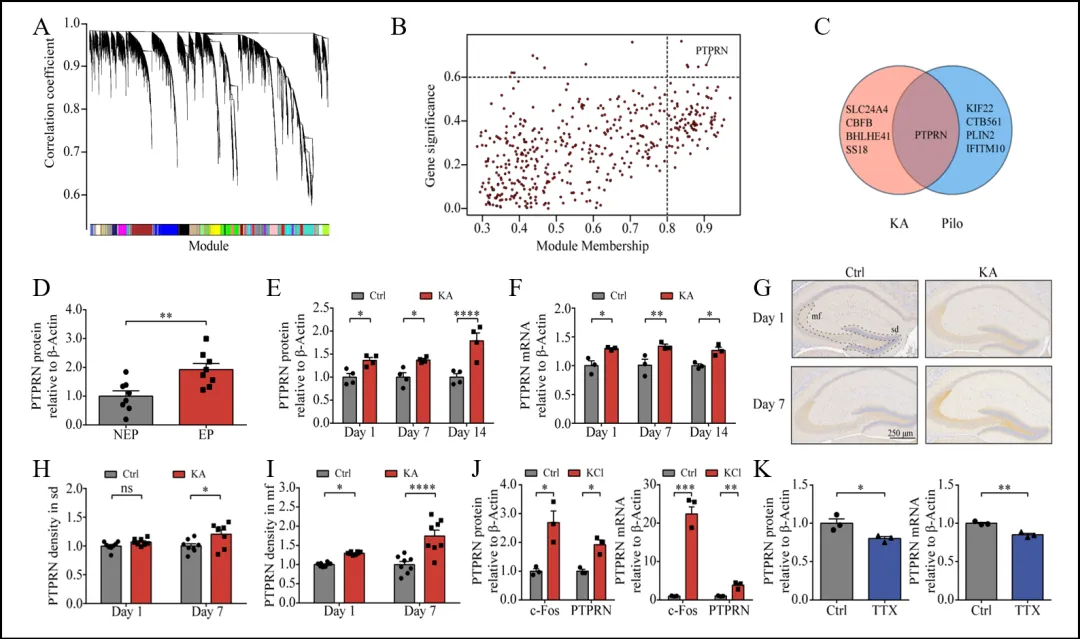
图1 PTPRN在TLE患者和动物模型中的表达水平异常升高
PTPRN是受体型蛋白酪氨酸磷酸酶(RPTPs)家族成员之一,其在调节胰岛素分泌中的作用已被广泛研究,但其在海马颗粒细胞中的功能尚不明确[7,8]。研究人员生成了PTPRN敲除(PTPRN-KO)小鼠,对其急性脑片进行全细胞电流钳记录(图2 A,B)。结果显示PTPRN-KO小鼠海马颗粒细胞动作电位(AP)数量显著增加,表明这些神经元的内在兴奋性增强。此外,PTPRN-KO小鼠的AP上升速度明显加快,反映神经元钠通道电流增强(图2 C-F)。在KA模型中,PTPRN-KO小鼠显示出增强的癫痫易感性(图2 K,L)。相反,PTPRN过表达(PTPRN-OE)小鼠的内在兴奋性降低,癫痫易感性显著降低(图3 G-J,O,P)。慢性癫痫模型中,PTPRN-KO小鼠表现出更早的自发性癫痫发作,并且慢性癫痫发作的严重程度显著增强(图2 M,N,Q,R)。这些结果表明,PTPRN通过降低神经元的内在兴奋性,抑制癫痫易感性和严重程度。

图2 PTPRN降低神经元内在兴奋性并抑制癫痫易感性
为了探究PTPRN抑制神经元内在兴奋性和癫痫易感性的分子机制,研究者进行了免疫沉淀-质谱分析(IP-MS),鉴定出与PTPRN相互作用的蛋白,其中包括NaV1.2钠通道(图3 A,B)。NaV1.2对调节神经元兴奋性至关重要[9]。免疫荧光和Co-IP分析显示,NaV1.2与PTPRN共定位于海马DG颗粒细胞中(图3 C-E)。通过全细胞膜片钳技术记录NaV1.2介导的电流,发现PTPRN显著减少了NaV1.2的电流密度,并导致激活曲线去极化偏移,表明PTPRN负向调节NaV1.2通道功能(图3 F,G)。此外,PTPRN对钠通道亚型表现出选择性,不调节NaV1.5和NaV1.6通道功能(图4 H,I)。
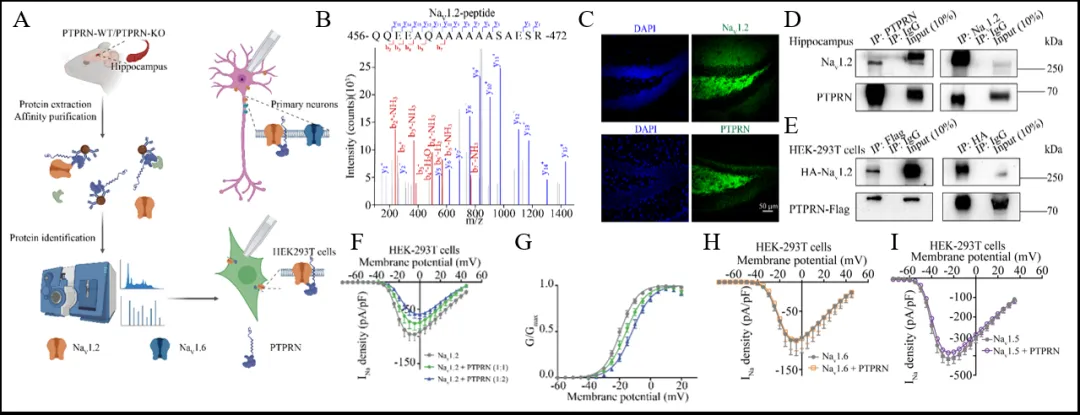
图3 PTPRN与NaV1.2通道相互作用并负向调控NaV1.2通道功能
PTPRN负向调节NaV1.2电流密度,提示PTPRN可能影响NaV1.2通道的表达或转运。为检测NaV1.2通道的表达和转运,研究者进行了细胞膜蛋白生物素化检测。结果显示,尽管NaV1.2的总蛋白水平不变,但敲低PTPRN显著增加了NaV1.2的细胞膜表达(图4 A,B)。对IP-MS的结果进行KEGG富集分析揭示,PTPRN的相互作用蛋白多数与内吞相关,提示PTPRN可能通过促进内吞来调节NaV1.2功能。在共表达NaV1.2和PTPRN的HEK-293T细胞中进行电生理记录,研究者使用内吞相关GTP酶Dynamin的抑制剂Dynasore,发现PTPRN对钠电流的抑制作用消失,证明PTPRN通过内吞调节NaV1.2通道定位(图4 C,D)。为了排除PTPRN影响NaV1.2细胞内运输的可能,研究者使用BFA来阻断蛋白从内质网到高尔基体的运输,发现未能消除PTPRN的抑制效应,表明PTPRN并不影响NaV1.2细胞内运输(图4 C,G)。使用Clathrin组装抑制剂Pitstop2,PTPRN对电流的抑制作用消失,表明Clathrin介导的内吞(CME)参与PTPRN对NaV1.2的调节(图4 C,H)。此外,应用E1泛素酶抑制剂TAK243和HECT E3泛素连接酶抑制剂Heclin均能消除PTPRN的抑制效应(图4 E,F),表明PTPRN通过促进泛素依赖性内吞作用调节NaV1.2通道功能。
为了进一步阐明PTPRN对NaV1.2选择性调控的机制,研究者构建了NaV1.2和NaV1.5的C末端交换嵌合体。结果表明,NaV1.2的C末端替换为NaV1.5的C末端后对PTPRN的调控产生抗性,而NaV1.5与NaV1.2的C末端融合后对PTPRN调控敏感(图4 I-K)。NaV1.2的C末端包含HECT E3泛素连接酶NEDD4和NEDD4L识别并结合的PPSY基序,突变NaV1.2的PPSY基序后,PTPRN引起的NaV1.2抑制效应完全消失,表明PPSY基序在PTPRN对NaV1.2的调控中起重要作用(图4 L)。Co-IP实验证明PTPRN在体内外与NEDD4L而非NEDD4相互作用(图4 O,P)。过表达失活的NEDD4L突变体或敲低NEDD4L均能消除PTPRN对NaV1.2的调控能力(图4 M,N),表明PTPRN通过促进NEDD4L介导的NaV1.2泛素化,调控NaV1.2通道功能。

图4 PTPRN通过促进NEDD4L介导的NaV1.2通道的泛素化和泛素依赖性内吞,调控NaV1.2通道的功能
为验证PTPRN-NaV1.2轴对神经元内在兴奋性和癫痫易感性的调控作用,研究团队在PTPRN-KO小鼠中敲低了NaV1.2表达。PTPRN缺失导致海马DG颗粒细胞内在兴奋性增加以及癫痫易感性增加,而敲低NaV1.2能够逆转这些变化(图5)。以上结果说明,降低PTPRN-KO小鼠中的NaV1.2表达可以逆转PTPRN缺失引起的异常内在可塑性和癫痫易感性。这一发现为理解PTPRN对NaV1.2通道功能的影响奠定了基础,并为开发精确调控NaV1.2通道的方法提供了依据。

图5 PTPRN-NaV1.2轴调节神经元内在兴奋性和癫痫易感性

图6 文章总结图
文章结论与讨论,启发与展望
综上所述,该研究结合生物信息学、分子生物学及电生理实验技术等手段,揭示了PTPRN在中枢神经系统中作为活动依赖性内在神经元可塑性的调节因子。神经元活动增强后,PTPRN表达增加,通过招募E3泛素连接酶NEDD4L促进NaV1.2钠通道泛素化和随后的内吞,从而抑制NaV1.2通道功能,降低细胞兴奋性,进而在过度兴奋的神经网络中发挥潜在的保护作用。
该研究提出的PTPRN-NaV1.2轴调节机制为神经网络活动的增强提供了反馈效应,从而帮助调节神经元的内在可塑性。对该机制的研究不仅为将来癫痫治疗方法的开发提供了理论依据,而且进一步完善了对神经系统兴奋性可塑性的理解。然而,该研究目前缺乏直接的临床证据,基于该机制的疗法有待进一步研究和验证。
专 家 点 评
许琪(教授,中国医学科学院基础医学研究所,国家杰出青年基金获得者)
药物治疗是癫痫患者的主要治疗手段,当前的抗癫痫药物主要通过调节中枢神经系统的兴奋-抑制平衡来发挥作用。然而,癫痫耐药一直是领域的一大难题,大约30%的癫痫患者对现有药物反应不佳,亟需进一步解析癫痫发病机制并探索新的药物干预策略。北京大学药学院黄卓教授与重庆医科大学附属第一医院田鑫教授团队的研究成果为耐药性癫痫的机制探究和药物开发提供了新方向。他们的研究首次揭示了PTPRN在中枢神经系统中作为活动依赖性内在神经元可塑性调节因子的角色,探究了通过内吞NaV1.2钠通道抑制其功能,降低神经元膜兴奋性进而改善癫痫发作的独特机制。这一发现不仅为未来癫痫干预靶点的开发提供理论基础,同时展示出了较好的临床转化潜力。同时,该研究还为其他神经系统相关疾病的治疗提供了新的启示和参考。
吴海涛(研究员,军事医学研究院,国家杰出青年基金获得者)
全球大约有5000万人深受癫痫疾病的困扰。虽然临床上有30余种抗癫痫药物,但仍有30%的癫痫病人药物治疗无效,究其原因在于导致癫痫发作的原因多样,且致病机制并未完全阐明,因此癫痫的治疗需要新靶点和新策略。近期,北京大学黄卓教授和重庆医科大学田鑫教授合作,首次鉴定出一种全新的泛素连接酶适体PTPRN(蛋白酪氨酸磷酸酶受体型N),其能够显著抑制难治性颞叶癫痫的发生和发展。这项研究创新性地将PTPRN作为泛素连接酶适体引入癫痫研究领域,揭示了其在神经元内在兴奋性调控中的重要作用。该研究为理解离子通道转运在癫痫发病中的作用机制提供了新的理论基础,并为未来研发基于NaV1.2通道功能调控的精准治疗策略提供了新思路。该发现不仅为理解癫痫发生的致病机制提供了新的实验证据,也为未来针对癫痫的新型药物研发和临床治疗提供了新思路。
本文共同第一作者为王一帆、杨辉、李娜、汪李丽。重庆医科大学附属第一医院田鑫教授、北京大学药学院黄卓教授为论文通讯作者。
参考文献
1.Oberman, L., & Pascual-Leone, A. (2013). Changes in plasticity across the lifespan: cause of disease and target for intervention. Progress in brain research, 207, 91–120.
2.Feldman D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annual review of neuroscience, 32, 33–55.
3.Debanne, D., Inglebert, Y., & Russier, M. (2019). Plasticity of intrinsic neuronal excitability. Current opinion in neurobiology, 54, 73–82.
4.Sehgal, M., Song, C., Ehlers, V. L., & Moyer, J. R., Jr (2013). Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiology of learning and memory, 105, 186–199.
5.Boison, D., & Steinhäuser, C. (2018). Epilepsy and astrocyte energy metabolism. Glia, 66(6), 1235–1243.
6.Xiao, K., Sun, Z., Jin, X., Ma, W., Song, Y., Lai, S., Chen, Q., Fan, M., Zhang, J., Yue, W., & Huang, Z. (2018). ERG3 potassium channel-mediated suppression of neuronal intrinsic excitability and prevention of seizure generation in mice. The Journal of physiology, 596(19), 4729–4752.
7.Lan, M. S., Lu, J., Goto, Y., & Notkins, A. L. (1994). Molecular cloning and identification of a receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA and cell biology, 13(5), 505–514.
8.Mziaut, H., Trajkovski, M., Kersting, S., Ehninger, A., Altkrüger, A., Lemaitre, R. P., Schmidt, D., Saeger, H. D., Lee, M. S., Drechsel, D. N., Müller, S., & Solimena, M. (2006). Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nature cell biology, 8(5), 435–445.
9.Spratt, P. W. E., Ben-Shalom, R., Keeshen, C. M., Burke, K. J., Jr, Clarkson, R. L., Sanders, S. J., & Bender, K. J. (2019). The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron, 103(4), 673–685.e5.
原文链接
http://doi.org/10.1002/advs.202400560